產品與服務
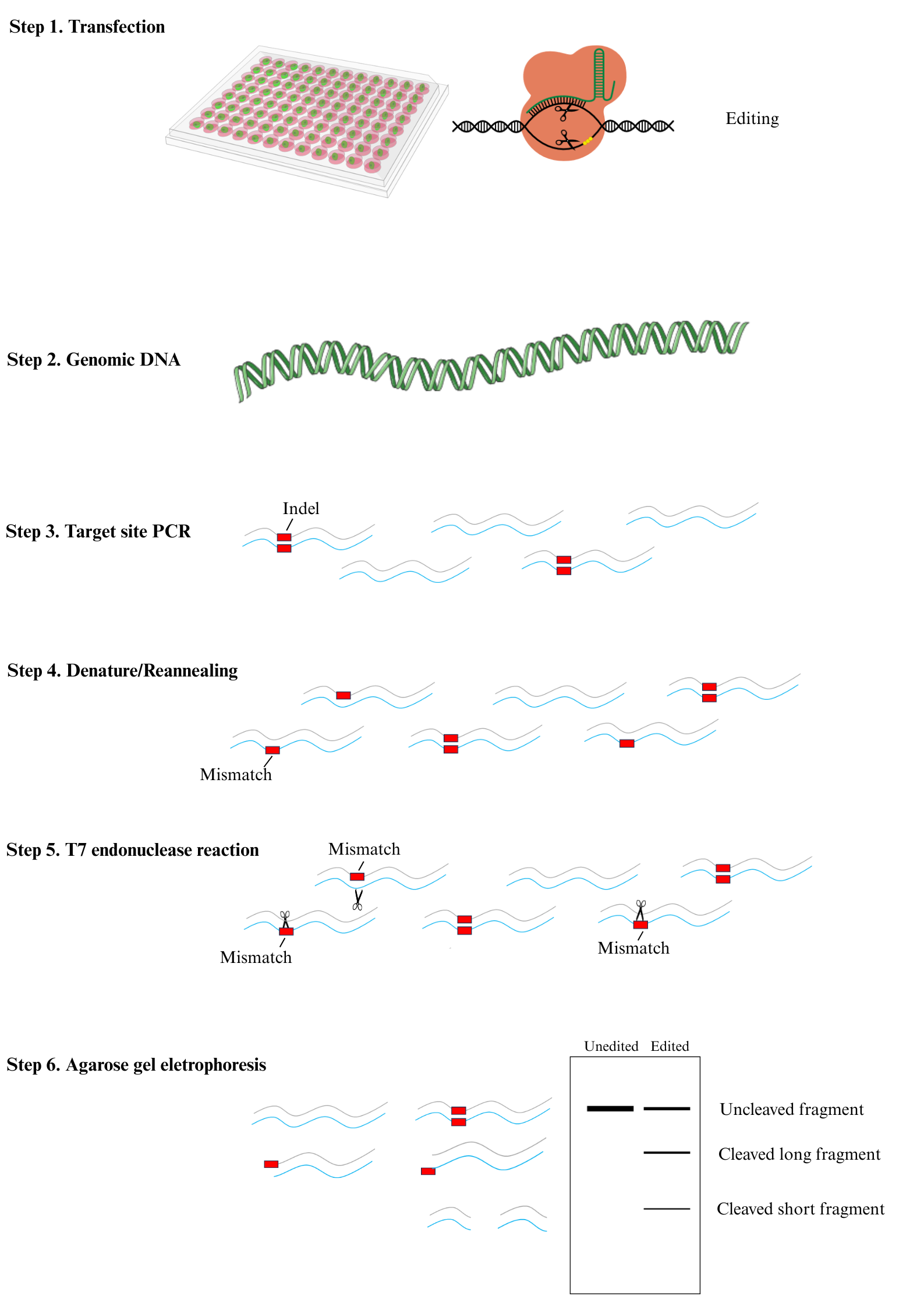
基因編輯突變檢測原理:
提取基因編輯后細胞的基因組DNA;采用高保真酶PCR擴增基因組DNA中被編輯位點附近的DNA片段;擴增的DNA片段進行變性和退火;最后用T7EI酶切鑒定。
編輯成功的細胞的基因組DNA會產生堿基插入或缺失突變,PCR擴增的DNA片段變性和退火后,未突變的和突變的基因之間,不同突變的基因之間,會形成不完全配對的雙鏈DNA,T7EI酶能識別并酶切不完全配對的雙鏈DNA。
T7EI酶切產物進行電泳分析,通過灰度值分析,即可獲得基因編輯效率。
| 分類 | 成分名稱 | 5T | 25T |
|---|---|---|---|
| Part I | Buffer GL1 | 1 mL | 5 mL |
| Buffer GL2 | 1 mL | 5 mL | |
| Buffer GW1 | 1.3 mL | 6.5 mL | |
| Buffer GW2 | 1.5 mL | 7.5 mL | |
| Nuclease-free Water | 1 mL | 5 mL | |
| DNA Collection Column | 5 T | 25 T | |
| Part II | Proteinase K | 100 μL | 500 μL |
| Control Template | 5 μL | 10 μL | |
| Control Primer Mix | 5 μL | 10 μL | |
| 2× High-fidelity PCR Premix | 50 μL | 250 μL | |
| T7 Endonuclease I (10 U/μl) | 5 μL | 25 μL | |
| 10× T7 buffer | 10 μL | 50 μL |
![]()
實驗流程
Part I常溫保存,Part II -20℃保存。
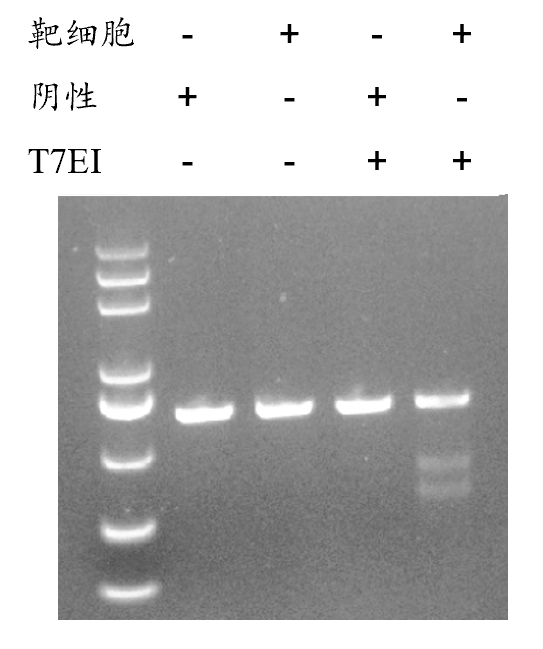
基因編輯效率= 被剪切條帶的灰度總和/ 剪切條帶和未被剪切的特異性條帶的灰度總和

